ゲノムの時代へ
膨大な情報を、
わたしたちは自在にあやつって、
新しい時代を拓くことができるのか、
やがてくる明日への羅針盤に
感染症の出口戦略
感染症に限らず、疾病対策の基本は費用対効果分析です。費用対効果分析とは、ある疾病を〇〇の程度減少させるためには、いったいいくらの費用がかかるのかを検討するもので、がん検診の対策立案などにも広く用いられています。費用対効果分析では「命」か「経済」かの二者択一を行うのではなく、ある効果を得ようとすると費用は必ずかかるものであるから、両者が最もバランスよく保たれるポイントを議論しようとしています。
ある疾患に関する公衆衛生学的対策の王道は以下のとおりです。
- リスクを予測する。個人のリスクおよびその疾患の広がりなどを含む
- ある対策にどれほどの効果があるのかを推定する
- 対策には常にコストや犠牲を伴うため、その対策の費用対効果を検証し、情報公開と透明な議論を経て実施有無の判断を行う
このように費用対効果分析は疾病対策の根幹を形成しています。
では費用対効果分析からみた具体的な出口戦略とはどのようなものでしょうか。まずは命、健康を守るという観点からは少なくとも以下の3つの指標を検討していかなければなりません。
1. 実効再生産数
「1人の感染者がその後何人の方に感染させるか」の指標です。1人を超えると感染者は増加し、1人を下回れば感染は終息に向かっていくと直感的に理解できます。この指標が「1」を下回ってくれば、その感染症の致死率にもよりますが、出口に近づいていると考えられます。
この実効再生産数はどのように算出されるのでしょうか。ある指標を算出するには通常、モデルというものを構築し、そこにパラメーターと呼ばれる数値を代入してその指標を導き出します。身近な例でいえば、天気予報では、数値予報モデルに世界中から送られてくる各地点の気圧、気温、風などの値を代入し、天気予報や警報の発出を行います。同じように感染症では、SEIRモデルというモデルが提唱されており、そこにパラメーターを代入し、1人の感染者がその後何人の方に感染させるのかの数値をはじき出しています。SEIRのSは感染症に対して免疫を持たない者(Susceptible)、Eは感染症が潜伏期間中の者(Exposed)、Iは発症者(Infectious)、Rは感染症から回復し免疫を獲得した者(Recovered)を指します。パラメーターとしては、以下の3つの数字が必要です。
1) 感染率
2) 潜伏期間
3) 感染期間
このパラメーターの中で、「感染率」が非常に重要な数値です。これをできるだけ正確に得るためには、感染の有無を調べるための十分な検査体制が整備され、その人が何人の方に感染させたかをつぶさに調べていくことが必要です。疾病対策の基本である実態把握のためには、当然ながら検査を受けた方の数と陽性の方の数を全国から集計し、迅速かつ正確に一元管理するシステムも必要です。未だ完備されていないのであれば、データ送信を電子化することも考慮すべきです。
また、モデルにはいくつもの種類があり、かつパラメーターも数値の採用に関していろいろな選択肢があります。その指標算出の仕方について、研究者コミュニティで議論していくことも大切な手続きと思われます。
2. 検査体制
これを整備することは感染症制御の基本です。検査をして陽性であれば、病院のみならずホテルなどもフル活用して隔離します。十分な検査体制が整備されていない状況では有病率や上記の実効再生産数などを十分な精度で算出することはできません。有病率に関しては陽性率などを代替指標として用いることもある程度は可能です。例えば、PCR検査の感度※が80%、特異度が99%、陽性率が10%である場合、有病率は0.14%と推定はできますが、感度は現時点でそれほど正確に算出されているものではなく、報告によって50%~80%くらいの幅があります。感度があやふやな段階で有病率を推定することは誤った数値を出してしまう危険があります。やはり1日当たりの検査実施数は必要かつ重要な指標の一つです。
※感度、特異度、陽性率などの用語については、日本疫学会の「新型コロナウイルス関連情報特設サイト」感染症疫学の用語解説のページの「7. 検査の正確さの指標」をご参照ください。
3. 医療体制
感染者が多くなった場合でも、重病者に対する医療体制がしっかり整備され、その受け入れ体制に余裕があれば、死亡者をある程度は減少させることができます。ある地域にはいくつの病院があり、そこには何床のベッドがあって何人の患者さんを受け入れることができるのかも、特に命を守るという観点から重要な指標です。
ここまでは命という観点からの指標をみてきました。ここからは、経済という視点からの指標をみていきます。
4. お金
感染症対策はその多くが人と人の接触を減少させる方向で実施されるものですから、どうしても経済的な損失が生じます。あまたの経済指標について、ある対策を実施した場合、どれくらいの時間が経過すると個々の指標がどれほど動くと予想されるのかを検討していくことが必要です。特に感染症では人と人が接触する可能性が高い業種ごとの経済指標に加えて、特定の業種が大きな痛手を負った場合に、マクロ経済にどのような影響があるのかも議論が必要です。対策立案には経済学者の参画は必須です。
5. もの
経済の柱のひとつである「ものづくり」にどのような影響があるのかをみなければなりません。自動車工業やパルプ・紙・紙加工品工業などの生産量はその対策によっていかほどの影響を受けているのか、食料品や日常品などはどうかなど、鉱工業指数や食料品工業指数やそれと類似した短期的な指標などのモニタリングが必要です。さらに直近の懸念としては、今必要とされているマスクや防護服などの医療や予防に使用されるべき「もの」について、供給が遅延するような事態を引き起こしていないかの確認が必要です。
6. ひと
医療関係者にかかる大きな負担は何らかの数値化を行い考慮すべきです。また、最も重要な「ひと」に関する指標のひとつには教育があります。学校を休校にするのであれば、教育という「ひと」への重要な投資が奪われるという費用(コスト)を考慮に入れなければなりません。家庭に閉じこもることで、虐待やDVなどの問題も深刻化するかもしれませんので対策が必要です。また、飲食業やホテル業などには人の心に栄養を与える効果があるのかもしれません。これを指標化するのはなかなか難しいですが、いずれにしても一律に不要不急だからいつまでも休業していいというものではないと思われます。それぞれの職業にはそれぞれの重要性があります。「ひと」を育てるあるいは癒すといった効果についても要検討とすべき指標です。これは特定の業種の収入減という指標とともに考慮されるべきものと思われます。
出口戦略の在り方
以上みてきたように、命に関しては1~3の指標を算出し、経済に関しては4~6の指標に鑑み、両者同時に並べて入口戦略と出口戦略を検討します。1~3の命に関する指標については、ひとつの対策が他の対策の効果を相乗的に上昇させることもありますし、経済に関する指標においても、失業・減収などから命や健康の問題にまた直結することもあります。要は1~6は常に有機的に影響し合っていることを念頭においておくことが必要です。また、行動変容には「脅し」手法はあまり有効ではなく、シンプルなメッセージと自然と入ってくるものである必要があることもこれまでの延々と続けられてきた公衆衛生対策で明らかとなっています。「人の行動はおいそれとは変わらない」ことを前提に対策を検討します。出口戦略としては、命に関する1~3の指標がすべてある閾値を下回るあるいは越えて青信号となり、かつ経済に関する4~6の指標すべてが赤信号になった場合には、おそらく躊躇なく出口である休業要請解除に進む判断が下されますが、現実には6つの指標が赤、黄色、青の入り混じった状態になります。指標群はこのような状態であるが、何を重視し、何を守ることが費用対効果の観点から最も賢明であるかを判断し、段階的に〇〇のような解除を行っていくという決定に至ります。この過程で重要なことは、それぞれの指標がどのように算出されてきたのか、出てきた指標群をどのように議論したのかを十分に公開し、さらに国民に向かって丁寧に説明していくことです。最終的にどのような出口戦略であっても、万人の利益が最大化されることはなかなか困難で、身体が弱い方にとっては早期の解除は命に係わると思われるでしょうし、休業要請が長引く業種の方々にとっては、経済的に命を失うと思われるでしょう。したがって、入口である対策の決定を行ったのであれば、そのことで相対的に大きな損失を被ることになる方々には公的資金で補償をするあるいは家庭内における虐待やDVに関する対策が必要ですし、出口においては命に関わるリスクが高くなる方々に、自治体の保健師さんなどがより活動しやすくなるような措置が必要です。費用対効果分析で全体の戦略を描いて実行した後は、そこで大きなしわ寄せを受ける方々に必要な施策を実施していくことが求められます。また、ワクチンや治療薬の開発が実現された場合、あるいは逆に感染の第2波等が襲来した場合には、すみやかにその時点の数値で費用対効果分析を行い、遅滞なく対策を柔軟に修正することが重要です。
公衆衛生対策を後押しするゲノム情報 -新型コロナウイルス感染症クラスター対策の視点から-
現在、世界中で流行が拡大している新型コロナウイルス感染症(Novel Coronavirus disease 2019: COVID-19)。その原因となるのが新型コロナウイルス (SARS-CoV-2) です。SARS-CoV-2を含むコロナウイルスは、外側を膜(エンベロープ)で覆っているエンベロープウイルスです。ウイルスのゲノムはRNAで、約3万塩基とRNAのゲノムを持つウイルスの中でも最長です。ウイルス粒子は直径 約100-200 nmで、S (スパイク)、M (マトリックス)、E (エンベロープ)という3つのタンパク質で構成されています。ウイルスは、相手に感染しようとするとき、その相手の細胞のとっかかりのようなもの(受容体)を見つけて、結合してから細胞に入り込みます。ウイルスの側で、その受容体と結合するのはSタンパク質です。Sタンパク質が鍵ならば、細胞の受容体が鍵穴になります*1。ヒトに感染するコロナウイルスとしては、風邪の原因ウイルスとしてヒトコロナウイルス229E、OC43、NL63、HKU-1の4種類、そして、重篤な肺炎を引き起こす2002年に発生した重症急性呼吸器症候群 (SARS)コロナウイルスと2012年に発生した中東呼吸器症候群 (MERS)コロナウイルスの2種類が知られています*2。SARS-CoV-2は、相同性約80%とSARSコロナウイルスに近く、SARSコロナウイルスと同じ受容体 (ACE2)を使ってヒトの細胞に吸着・侵入することが最近の研究で報告されています*1。
日本においては、2019年末の中国・武漢を発端とするSARS-CoV-2が2020年1⽉から2⽉にかけて国内に流入し、地域的な感染クラスター(集団)を発⽣させました。クラスターが発⽣した場所では「積極的疫学調査」(感染症などの色々な病気について、発生した集団感染の全体像や病気の特徴などを調べることで、今後の感染拡大防止対策に用いることを目的として行われる調査)が実施され、発⽣源と濃厚接触者を特定することで、さらなる感染拡⼤を封じ込める対策が展開されてきました。一時的に⼀定の成果を得た地域もありますが、後に各地で感染拡⼤が進⾏し、2020年4月28日現在で全国規模の緊急事態宣⾔に⾄っています。
感染拡大の状況を把握し、今後の対策を立てる上で積極的疫学調査は重要ですが、それを裏付け科学的に支援するのがゲノム情報です。世界各地の研究所においてSARS-CoV-2のゲノム配列が解読されており、4月16日現在で4,511人から採取した検体のSARS-CoV-2ゲノム配列がGlobal lnitiative on Sharing All Influenza Data:GISAIDのデータベースに登録されています。日本国内でもSARS-CoV-2のゲノム解読は進められています。4月27日、国立感染症研究所からSARS-CoV-2に関する分子疫学調査の結果が発表*3されました。日本国内で各地の協力施設から収集された陽性者の検体を用いて562患者でのゲノム解読が行われ、世界のデータと統合した塩基変異抽出、ウイルス株の親子関係を示すハプロタイプネットワーク図が作成され、その分析結果が解説されています。SARS-CoV-2の変異速度は現在のところ25.9塩基変異/ゲノム/年(つまり、1年間で25.9箇所の変異が⾒込まれる)と推定*4されています。2019年末の発⽣から4ヶ⽉ほどの期間を経て、ゲノム全域に少なくとも9塩基ほどの変異がランダムに発⽣していると⽰唆されています。
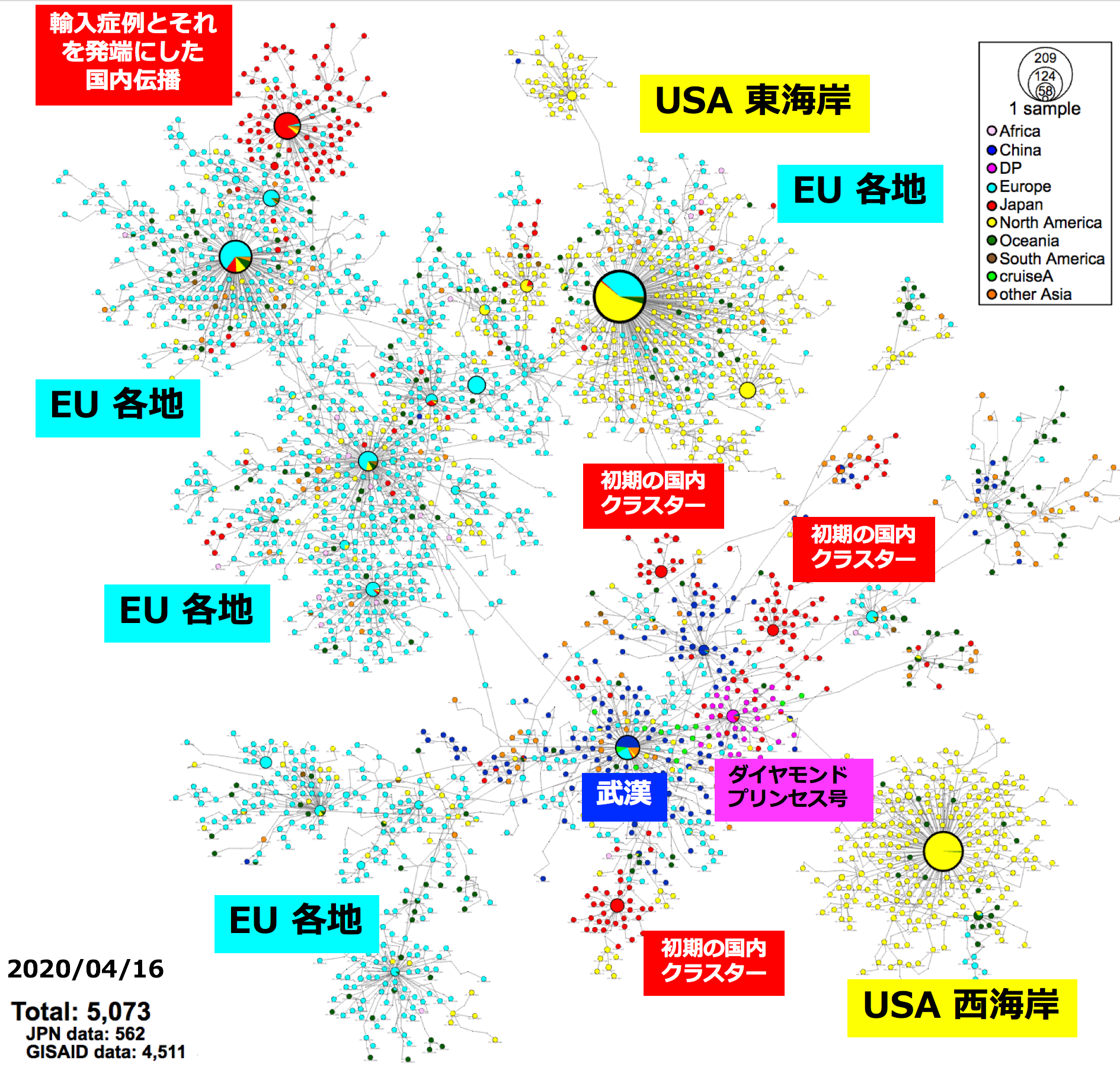
図:世界と⽇本のSARS-CoV-2ゲノム情報の塩基変異を⽤いたハプロタイプネットワーク図(参考文献3より)
日本では、中国発の第1波においては “中国、湖北省、武漢” をキーワードに感染者を特定し、濃厚接触者をいち早く探知して抑え込むことができていました。しかし、緻密な疫学調査により収束へと導くことができるかと思われた矢先に、3⽉中旬から全国各地で “感染経路(リンク)不明” の孤発例が検出されはじめています。渡航⾃粛が始まる3⽉中旬までに海外からの帰国者経由で “第2波” の流⼊を許し、数週間のうちに全国各地へ伝播して “渡航歴なし・リンク不明” の患者・無症状病原体保有者が増加したこと、3⽉中旬以降、国内移動や外出自粛などの⾏動制限の徹底がなかなか出来なかったことで、SARS-CoV-2が国内に徐々に広まり、現在の感染拡⼤へ繋がったと推測されています。今後、第3波、第4波が来ることは十分に考えられます。ゲノム情報と疫学調査情報の迅速な共有、公開が効果的な感染症対策の鍵と言えるでしょう。
参考文献
1. 日本ウイルス学会「新型コロナウイルス感染症について」
2. Cui, J., Li, F., Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019. 17181-192
3. 国立感染症研究所病原体ゲノム解析研究センター. 「新型コロナウイルスSARS-CoV-2のゲノム分子疫学調査」
4. 変異速度の推定値は、最新の成果をもとに随時更新されています。最新のデータは、Nextstrain等をご参照下さい。
“スティグマ”を避けるには~感染症の流行で、そしてゲノム研究でも
「スティグマを避けよう」という言葉、聞いたことがありますか?
新型コロナウイルスの感染拡大を受けて、スティグマを避けよう、という声が上がっています。“スティグマ”、日常ではあまり聞きなれない言葉ですが、英単語の直訳では、汚名・烙印といった意味になります。もともとは奴隷や家畜に押された焼印に由来し、キリストが十字架で受けた傷あとなども指す言葉とされます。
さて、現在、問題にされているのは社会的スティグマです。社会において、何らかの属性を持つことから差別や偏見の対象として扱われてしまうようになることを指します。属性は、民族や外見、障害などであることもあり、あるいは、何らかの疾患の罹患ということもあります。
本稿執筆現在(2020年4月23日)、新型コロナウイルスの感染拡大の局面で、問題になっているのは、ウイルス感染や、それに少しでも関係することが、そのスティグマの対象となっている事態が懸念されていることです。実際、感染者を出した機関の職員等が、各種サービスの利用を拒絶されたりする、といった事態が発生しているという報道があります。保育園利用や入店を断るといった対応は、相手を根拠なしに接触するべきでないとするというスティグマ=烙印を押していることになります。こうした差別的な扱いがあってはなりません。
また、スティグマを避けることは、差別があってはならないということに加えて、寧ろ感染拡大を防ぐためにも重要になります。不必要に感染者を叩くようなことは、人々が感染について明らかにしづらくなったり、感染経路について秘匿するようになったりといったことに繋がります。
実はゲノム研究においても、スティグマを避けることは大きなテーマであり続けています。特定の家系から特定の疾患が多くあらわれる、特定の地域で遺伝性の疾患が多い傾向がある、といったことは、科学的事実であっても、そうしたスティグマに結びつきやすいものです。研究に際して、また、発表に際して、さまざまな配慮を行って、科学的な解明と、社会的な行動等との負の結びつきを断っていこうという努力がなされています。
今回の事態においても、それぞれの立場で、スティグマを避けていく努力が必要です。私たちは常に、感染拡大を防ぐ、という名目のもとに、何かの属性をもった人たちを苦しめることになっていないか、留意していく必要があります。
稀少疾患の原因探索に使われる日本人全ゲノムのバリアント頻度パネル
図を拡大
 ヒトはおよそ30億塩基対の配列をもっています。そのうち、たとえば一個人を見ると、常染色体で平均約420万箇所の塩基が、国際的な参照用配列に比べて異なります。その異なる部分をバリアントと呼び、集団が異なると、バリアントの組み合わせも異なることが分かっています。このバリアントの頻度について、世界的にいくつかの民族集団で調査が行われており、民族集団毎(例えばアフリカ系、東アジア系など)にまとめたデータベースが存在します。gnomADデータベースはその代表的なものの一つで、全ゲノムでは約1.5万人、遺伝子領域に絞ったもの(エクソーム)では約12.6万人を対象とした大規模データベースとなっています。
ヒトはおよそ30億塩基対の配列をもっています。そのうち、たとえば一個人を見ると、常染色体で平均約420万箇所の塩基が、国際的な参照用配列に比べて異なります。その異なる部分をバリアントと呼び、集団が異なると、バリアントの組み合わせも異なることが分かっています。このバリアントの頻度について、世界的にいくつかの民族集団で調査が行われており、民族集団毎(例えばアフリカ系、東アジア系など)にまとめたデータベースが存在します。gnomADデータベースはその代表的なものの一つで、全ゲノムでは約1.5万人、遺伝子領域に絞ったもの(エクソーム)では約12.6万人を対象とした大規模データベースとなっています。
一方、原因のよくわかっていない、稀な病気の要因を探るための遺伝子解析をする機会が現在増えつつあります。例えば、とある症状の方の遺伝子解析を行った結果、遺伝子領域で数万箇所のバリアントが検出されたとします。さて、この中から病気の原因になるバリアントはあるでしょうか、というのが解析目的のひとつです。原因バリアントがわかると、そのバリアントが属する遺伝子の機能や、バリアントに起因するタンパク質の機能変化などから病気の遺伝的背景が明らかになり、効果的な治療法を選択する足がかりになるからです。実際に、診断の難しい患者さんに対して、家族性も考慮して遺伝的要因を探る試みが日本で行われています (未診断疾患イニシアチブ IRUD)。
では、この病気の要因を探る遺伝子解析で、実際には数万の候補のほとんど(あるいはすべて)が、対象の病気と直接関係がないバリアントであろうことが想定される中、どのように原因バリアントを探すのでしょうか。最終候補へと絞り込んでいくその際に利用されるのが、バリアントを集めた大規模データベースなのです。特に稀な病気の場合は、集団である程度の頻度でみられるバリアントは、特定の病気と関連づけられないと推定されます。このことから、数万の病気の原因バリアント候補のうち、集団である程度(例えば1%以上)の頻度で見られるバリアントをふるいおとせるのです。
ただ人類の歴史的な背景から、民族集団によってバリアント頻度は異なり、特定の民族集団でのみ多くみられるバリアントは多々あるのです。したがって、解析対象の方が日本人であれば、日本人集団の頻度データを用いることで、より絞り込みの精度が上がることが考えられます。しかしながら、世界最大規模のデータベースgnomADにおいて、日本人由来の数は約80人程度と少ない状況です。東北大学東北メディカル・メガバンク機構(ToMMo)では日本人を対象としたバリアントデータベースを公開しており、2019年には対象を約4,700人に増やした全ゲノムのバリアント頻度パネル(4.7KJPN)を公開しました。ToMMoのバリアント頻度パネルは、これまで国内の各種の疾患研究において原因バリアントの絞り込みに活用されてきています。このように、病気の原因バリアントの同定に、住民コホートから得られたバリアント頻度パネルは重要な役割を果たすことができるのです。
【関連リンク】

各国ですすむ基準ゲノム構築プロジェクト
2019年2月、日本人基準ゲノム配列の初版JG1が公開されました。これは、1980年代に開始され2001年にドラフト配列決定に至った国際ヒトゲノム計画を、日本人検体を用いて再度実行したことに相当します。国際ヒトゲノム計画で構築された国際基準ゲノム配列(参照配列とも呼ばれる)は現代のゲノム解析に欠かせないものです。しかし国際基準ゲノム配列はもっぱらヨーロッパ系とアフリカ系に由来する検体から構築されたために、異なる民族集団に属する検体のゲノム解析には必ずしも適していませんでした。ゲノムのバリアントの検出は、解析対象のゲノム配列と基準ゲノム配列との比較により行われるのですが、解析対象の検体のゲノムを他の民族からの基準ゲノムと比較した場合、バリアントの見落としや誤検出が生じうることが分かっています。日本人検体から構築されたJG1を使用すると、日本人を対象としたゲノム解析において、これまで民族集団の違いに起因する見落としや誤検出を減らすことができるので、これまで未特定だった遺伝性疾患の原因究明や、がんを引き起こす遺伝子の同定などが期待されます。
JG1のように民族集団固有の基準ゲノム配列を構築する試みは各国で行われはじめています。しかしJG1は3人のゲノムを統合することでレアな配列バリエーションを可能な限り除外した点、および、遺伝地図等の情報を用いて解読した配列と染色体との対応づけも行った点が特筆に値します。例えば韓国ではAK1、中国ではHX1、またスウェーデンではSwe1, Swe2という高品質のゲノム配列が構築され報告されています。しかしこれらはいずれも1人のゲノム配列を決定したものであり、その個人がたまたま持っていたレアバリアントが基準ゲノム内に残ってしまっています。また手間のかかる染色体との対応づけは行わずに、数千本の配列の集まりとして公開されています。韓国からはKOREFという基準ゲノム配列も公開されていますが、こちらは染色体との対応づけの際に国際基準ゲノム配列の情報を利用しています。JG1は3人のゲノムを統合する前のどれか一つを見ても、各国のアセンブリよりも高い配列連続性を示していましたので、JG1は民族集団固有の基準ゲノム配列として群を抜いて高品質なものであると言えます。
2019年1月に米国では、350人の高品質ゲノム配列を構築するための研究の公募が発表されました。今後様々な民族集団の基準ゲノム配列が構築されること、さらにそれらの多様性を包括的に表現する方法の研究が進むと予想されます。そうすると民族集団ごとに構築した基準ゲノム配列を用いてより精度の高い解析が可能になるでしょう。さらにその次の段階はどうなるでしょうか?究極的には個々人のゲノム解読にも今回の基準ゲノム配列の構築で行われた様な手法による全ゲノム配列決定が利用されると予想します。
例えば、がんの解析では正常組織から抽出したDNAをもとに基準ゲノム配列を構築して、がん組織のバリアントを検出すればより精度の高い解析が可能になります。小児遺伝性疾患でも父・母・子で全ゲノム配列決定すれば、検出されたバリアントが祖先から受け継がれたものか、それともこの親子間で初めて生じた新生突然変異なのかの区別がより精度よく行われます。現在では夢物語とも言える解析ですが、DNA解読技術や情報解析技術の進展スピードを見ると案外近いうちに実現するかもしれません。
【関連リンク】
「日本人基準ゲノム配列」初版JG1の公開~日本人のゲノム解析がこれまでよりも精密かつ正確に~【プレスリリース】
最新記事一覧
-
- きこえと遺伝子医療の現在
- 2025.12.26|宇佐美真一
-
- 人体を取りまく常在菌〜機能を知って共存しよう〜
- 2020.12.16|後藤まき
-
- なぜウイルスははやく変化するのか?
- 2020.09.08|山口由美
-
- 棒状で増やすか環状で増やすか―新たなシークエンサー開発―
- 2020.07.16|川嶋順子
-
- 新型コロナウイルスに対する抗体
- 2020.05.18|峯岸直子