The linkage disequilibrium map (ToMMo_LD_map_192v1) has been released
Tohoku Medical Megabank Organization (ToMMo) constructed a linkage disequilibrium map (ToMMo_LD_map_192v1) using the whole-genome variation information from the Birth and Three-Generation Cohort Study (TMM BirThree Cohort Study), and we made it available widely on our website “jMorp”.
The linkage disequilibrium map by ToMMo is created from the whole-genome sequence of 96 people (192 haploids) of the members on the third generation among the family that participated in the TMM BirThree Cohort Study.
When a genome is inherited from a parent to the child, the information is mixed with those from their mother and father. There had been no accurate and large-scale data, which part of the chromosome is linked to other part or conversely, which is easily broken.
This ease of linking and broken is the genetic “distance” between each loci on the chromosome, but it is not necessarily same as the physical distance of how many bases are actually separated.
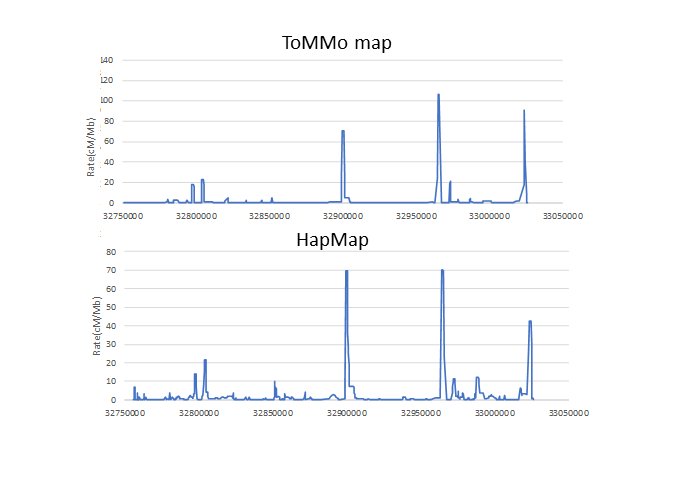
Linkage disequilibrium map is a kind of a map on human genome which expresses the ease of linkage and broken on the whole genome scale. So far, these kinds of maps have been created only for International HapMap Project and 1000 Genomes Project in the world.
This is the first release of a linkage disequilibrium map using Japanese families, and accurately generated data from deeply determined whole-genome sequences. The map will contribute to a wide variety of research from medical genetics such as fine-mapping of the disease genes, or a precise genetic imputation to evolutionary research.
We will increase the number of haploid genomes to be used and gradually upgrade to map with higher accuracy and resolution.
Release details
Linkage disequilibrium map
・ Genetic distance r (cM) list converted from the fine scale recombination rate σ on a population basis estimated from haplotype diversity data to a pedigree scale.
Abstract
‐Number of haploid genomes used: 192
‐Genome analysis: Whole genome analysis performed at Illumina Hiseq 2500 or Novaseq at an average depth of 25-30
‐Family: Three generation family, hepta or octa family
‐Phase determination: Phase determination was performed using shapeit2 + duohmm software from three generations pedigrees. This method is known to have higher phase determination accuracy than Merlin, the golden standard for family phase determination.
‐Genetic distance estimation: The fine scale recombination rate ρ is estimated from haplotype diversity data for 96 people using LDhat. The value divided by the regression coefficient to this deCODE recombination map corresponds the per-generation genetic distance on the pedigree scale.
Characteristic
-First calculated from the family of a Japanese ethnic group.
-The first estimation at the deep genome sequence level.
-The phase determination is accurate because the pedigree information used for the determination is abundant.
-Genotypic errors are efficiently removed using pedigree.
-Using an unprecedented number of haploid genomes.
Download
Please refer to the link below.
https://jmorp.megabank.tohoku.ac.jp/downloads
Related Link
