Estimating carrier frequencies of congenital metabolic disorders among 3,552 general Japanese individuals
Summary
- ・We estimated the carrier frequencies for recessive disorders from genomic data for the first time, using a whole-genome reference panel for Japanese people.
- ・We conducted an estimate on 32 causative genes of 17 congenital metabolic disorders, which are primary targets in newborn screenings in Japan. The estimated carrier frequencies for phenylketonuria and CPT2 deficiency based on variant interpretation guideline showed closer estimates to that by the reported incidence rates of the diseases. Those suggest that an estimation based on genomic diversity is possible.
- ・However, the results of comparisons of carrier frequency estimates from genomes and the reported incidence rates differed among eleven diseases with a single causative gene. And there may be possible factors contributing to this difference, such as inclusion of variants of mild effects or false positives, or presence of undetected pathogenic variants.
- ・Further elucidation of these factors by expanding data and revised methods for variant interpretation is expected to play a role in using genomic data to estimate the proportion of potential patients with recessive disorders in the general population.
Outline
Genetic disorders are categorized into several types, as known as “mode of inheritance”, depending on how the traits appear and how affected individuals appear in families. Humans have two copies of disease-causing genes, since each person receives two sets of genetic information from their parents. Dominant disorders occur if there is even one copy of the gene with a disease-causing variant. On the other hand, recessive disorders only occur when there is no copy that exerts the original function of the gene, and manifest when both two copies have the causative variants. The condition that a person has only one gene copy with a causing variant is termed a “carrier.” However, it is not yet clearly understood what proportion of general individuals have the causative variant as carriers. For single-gene disorders with only one causative gene, carrier frequency can be theoretically calculated, if the incidence rate of the disease is available. But in fact it has been difficult to elucidate what proportion of general individuals have a causative variant for a recessive disorder.
Tohoku Medical Megabank Organization (ToMMo) conducts large-scale sequencing of the whole genome of the Japanese population, and has constructed a whole-genome reference panel (At the time of article publication, consisting of 3,552 people, termed 3.5KJPNv2), and has released information on the frequency of genomic variants in Japan. This panel contains data on what proportion each genomic variant exist in the Japanese population. In this study, we used 3.5KJPNv2 (the version of whole-genome reference panel released in 2018, now the panel is expanded) data to investigate to what extent genomic diversity in a population can estimate the prevalence of recessive disorders. Specifically, we detected reported-and predicted-pathogenic variants in causative genes for congenital metabolic disorders, which are recessive disorders, and estimated the frequency of carriers. We then compared the results to the reported incidence rates, as based on the percentage of positive cases among the total number of people tested.
The genetic disorders that we focused on in this study are 17 congenital metabolic disorders for which causative genes are known, such as phenylketonuria, and are recessive disorders. These diseases are high priority for newborns screening due to the availability of treatment and the high accuracy of biochemical tests. Thus, many newborns are tested and statistics of incidence rates of each disease are highly accurate.
In this study, we analyzed genomic variants in 32 genes that are known to cause the 17 diseases. We performed biological and medical annotations, analyzed variants for all of the 3.5KJPNv2 variants, and detected reported-and-predicted pathogenic variants of the 32 genes using four different inclusion criteria. The frequency of carriers was estimated using the detected pathogenic variants and their frequencies. For comparison, we theoretically obtained the frequency of carriers from the reported incidence rates in past newborn screenings in Japan. The obtained values of carrier frequency based on genomic data were close to those estimated from the reported incidence rates in enzyme deficiencies, such as phenylketonuria and CPT2 deficiency. This suggests that it may be possible to estimate disease prevalence using genomic data.
However, the comparison of carrier frequencies, as estimated from genomic data and the reported incidence rates, varied from disease to disease. The differences in the estimated values may be due to various factors. For instance, one of possible causes of higher estimated values from the genome is inclusion of variants with mild effects, incomplete penetrance or false positives. On the other hand, lower estimated values from the genome indicate that undetected additional pathogenic variants exist. As research progresses further in the future, it is essential to take particular note of the following: 1) Additional line of evidence will increase the number of pathogenic variants; 2) the actual probability of manifestation of disease phenotype for each pathogenic variant remains unclear; 3) Common variants may contribute to pathogenesis in coexistence with other pathogenic variants, which may affect the proportion of affected individuals in a given population.
This study was the first initiative to show how genomic diversity in the human population could explain the prevalence of recessive disorders. By clarifying the direction and extent of the differences in the estimated carrier frequency, as based on both genome data and incidence rate, we can better understand the relationship between genomic variant frequencies and the percentage of potential patients in the general population.
[Figure 1: Outline of analysis in this study]
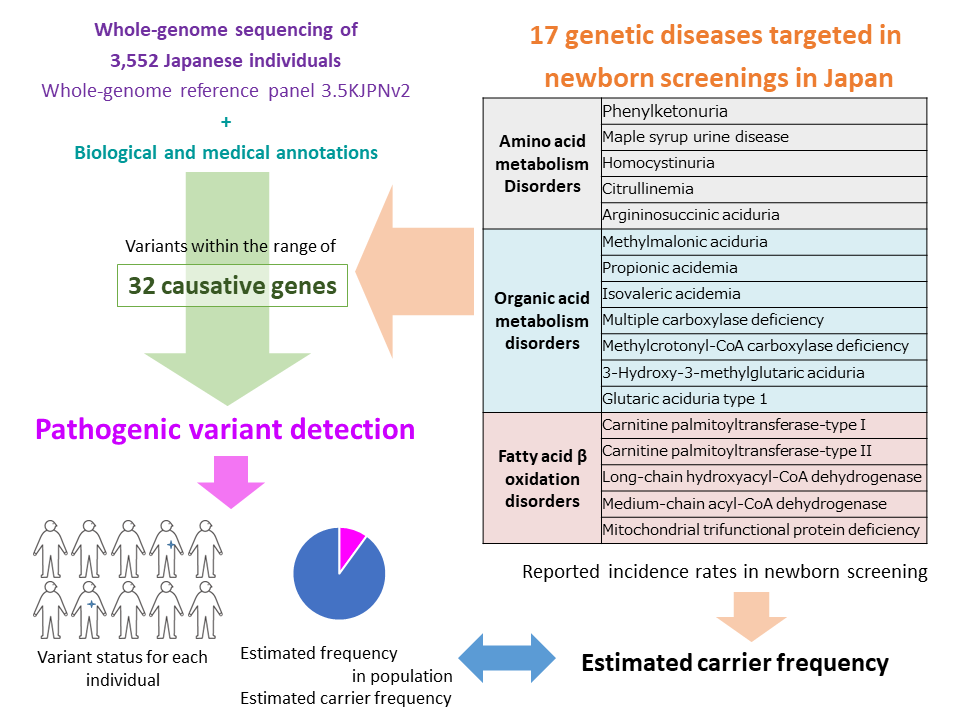
Medical and biological annotations were performed for all variants in the whole-genome reference panel 3.5KJPNv2. Further, the 32 genes that are responsible for diseases, which are primary targets for newborn screenings in Japan, were analyzed. Pathogenic variants were extracted using four different inclusion criteria (from a set of variants with a likely pathological significance to a set that contains potential candidates with pathological significance). Population frequency of risk alleles and carrier frequency were estimated based on the detected pathogenic variants.
[Figure 2: Comparison of estimated carrier frequency]
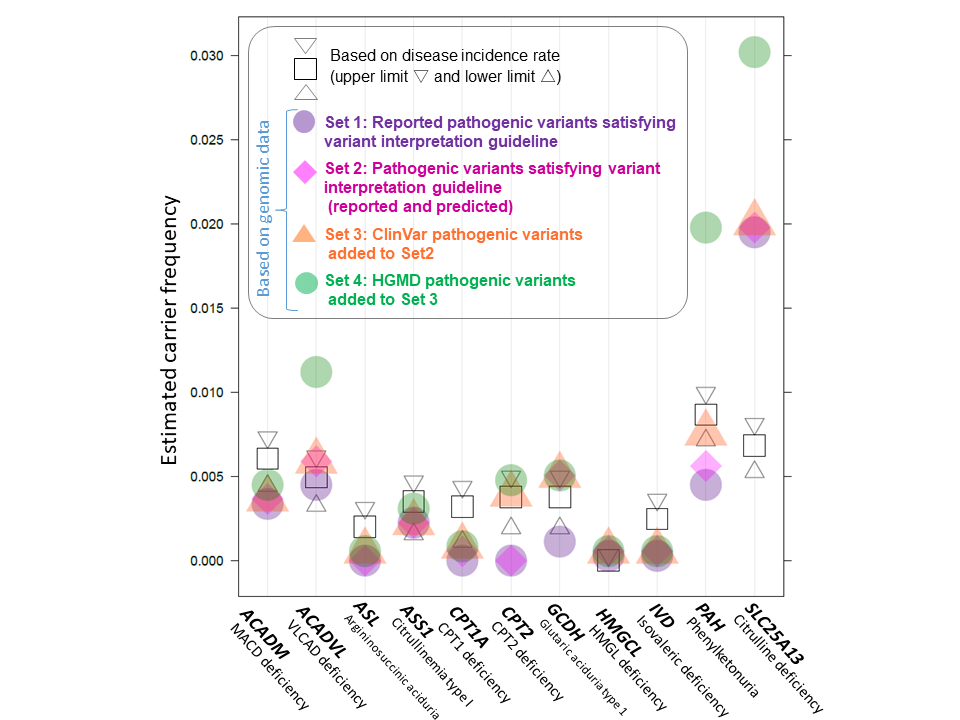
Carrier frequencies estimated from genomic variant data and that based on the reported incidence rates for 11 diseases with one causative gene.
Article information
Title: Estimating carrier frequencies of newborn screening disorders using a whole-genome reference panel of 3,552 Japanese individuals
Publishing journal: Human Genetics
Publish date: 18 March 2019
Authors: Yumi Yamaguchi-Kabata, Jun Yasuda, Akira Uruno, Kazuro Shimokawa, Seizo Koshiba, Yoichi Suzuki, Nobuo Fuse, Hiroshi Kawame, Shu Tadaka, Masao Nagasaki, Kaname Kojima, Fumiki Katsuoka, Kazuki Kumada, Osamu Tanabe, Gen Tamiya, Nobuo Yaegashi, the Tohoku Medical Megabank Project Study Group, Kengo Kinoshita, Masayuki Yamamoto, Shigeo Kure
doi: 10.1007/s00439-019-01998-7